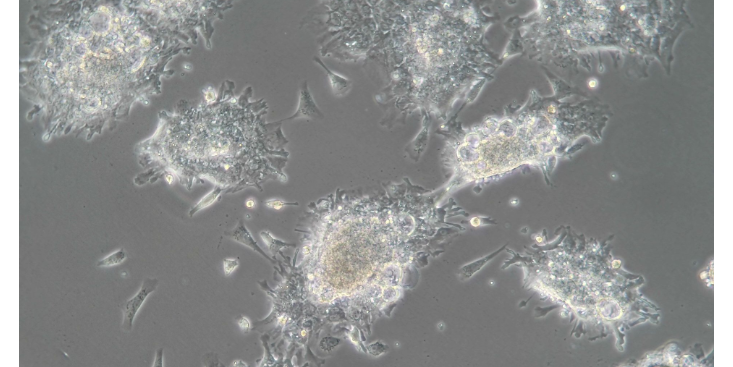
注意事项1.病毒包装的几个关键点主要包括:细胞因素、载体系统(尽量使用成熟的商业化载体系统)、构建重组的质粒正确与否、质粒抽提纯化情况、包装转染控制(24、48小时的细胞及荧光状态判断)、目的基因对病毒包装影响(基因大小、序列情况、蛋白功能毒性等都会影响到是否能包装成功)。,需要观察包装病毒后的48h培养基颜色是否橙红。3.病毒浓缩:病毒一般在48h和72h各收一次。如果不想浓缩病毒的话,也可以直接将收集的病毒上清作为要的细胞的培养基,但是可能效果会不太好。并且一般收病毒时,培养基的营养已经损耗了很多,那样直接培养细胞会损害细胞,所以建议还是进行浓缩后再。常见问题1.包装病毒时293T细胞状态不好,或者铺得过密,可以选择放弃该次实验。2.目的载体过大,不易。3.避免转染过程以及后续过程出现的污染。可以帮助科研人员深入理解疾病的共同性,即不同物种之间存在的共有病理变化过程。浙江血液科研技术服务购买
m6A研究又有新手段了?赶紧来了解一下吧!栏目:研究动态发布时间:2019-08-14RNA甲基化m6A是如今的研究热点之一,Cell上新发表了一篇介绍不使用m6A抗体的检测mRNAm6A水平的Resource文章......RNA甲基化m6A是如今的研究热点之一,目前主要的研究思路包括差异表达的write,reader和eraser基因分析;m6A抗体检测全转录组甲基化水平;分析m6A甲基化水平变化的靶基因和下游机制研究。而在7月25日的Cell上新发表了一篇介绍不使用m6A抗体的检测mRNAm6A水平的Resource文章,小编时间和大家分享一下。作者开发这一方法的前提是有研究报道了MazF能特异性的识别无修饰的ACA序列并发挥RNA酶活性在ACA之前剪切底物。但ACA序列的个A发生m6A甲基化之后将无法被MazF所识别,如图一所示。图1MazFRNA酶作用示意图根据这一原理,作者将纯化后的mRNA进行MazF酶处理,然后再对打断的RNA进行逆转建库测序。对于无修饰的位点,ACA处被完全剪切,测序reads正好分布于该位点上下游而且比对至该处的上下游reads数是相同的.如图2所示。而甲基化位点的reads会包含上下游的序列。作者开发了MAZTER-MINE软件包专门进行分析(图3)。海南外包科研技术服务实验科普知识 —了解IgM、IgG、IgA、IgE四种抗体。
1.首要原则:细胞不重要情况下立即丢弃,培养箱灭菌,所用培养基也都要丢弃,器械等重新灭菌或拆用新的。2.细菌污染一般都救不回来了,发现的时候培养基一般都很浑浊且细胞都死了3.污染且细胞很重要时:遇到念球菌污染,且细胞为基因改造细胞,非常重要。如231贴壁乳腺细胞,发现细胞周围出现很小的串珠透亮圆点,非常像念球菌污染,此时细胞状态尚可,且污染少。处理如下:用预热或室温PBS清洗3次,可适当振摇,将污染冲洗下来。随后加入10-20%双抗到培养瓶,置于37度培养箱1h,之后再用PBS清洗三遍,直至视野下无可见污染。此时细胞也被冲下大部分,因此此方法只适用在细胞贴壁强,状态好,密度高时使用。之后每天再更换培养基,每次用PBS冲洗2遍。过几天细胞状态尚可时,消化离心时用500r,3min,去掉上清,重复3次。这个方法是根据文献可利用念球菌和细胞体积重量差异实现分离。基本上这一步做完以后,污染就基本了,接下来就注意多观察,勤换液就行。
METTL3能够促进肺腺细胞的生长、生存和侵袭,但还不清楚它是否作为m6A调节器或效应器发挥作用[25]。在急性髓细胞白血病(AML)患者中,m(6)A调控基因的突变或拷贝数变化与TP53突变存在密切联系,且m(6)A调控基因的改变与AML不良预相关[26]。此外,FTO在AML中高表达,它通过降低mRNA转录本中的m(6)水平,调节ASB2和RARA等靶点的表达,增强了白血病基因介导的细胞转化和白血病形成,并抑制全反式维甲酸(ATRA)诱导的AML细胞分化[27]。在脂肪形成过程中,FTO表达与m6A水平成负相关,促进脂肪形成[3]。在胶质细胞瘤样细胞中,ALKBH5通过lncRNAFOXM1介导FOXM1基因pre-mRNA上的m6A修饰维持胶质瘤细胞的成瘤性[28]。此外,甲基转移酶METTL3或METTL14的敲除,能够改变m6A的富集和ADAM19的表达,极大地促进了胶质瘤细胞的生长、自我更新和形成[29]。图2m6ARNA修饰和介导的功能[30]m6A的研究方向主要是通过研究m6A修饰相关的甲基化、去甲基化酶和识别蛋白的功能,进而研究m6A修饰的生物学功能和作用机制:一般通过敲除m6A酶分子,研究下游功能基因分子的表达和m6A甲基化情况,通过介导相关基因异常(可变剪切、稳定性、翻译、miRNA调控)影响细胞表型和功能特征。动物疾病模型是一种用于研究人类疾病的重要工具。

二甲苯Ⅰ、Ⅱ各15min至透明。(2)放入二甲苯和石蜡等量混合液处理15min,再放入石蜡Ⅰ、Ⅱ透蜡各50~60min。透蜡在恒温箱内进行,箱内温度保持在55~60℃左右。4、包埋(1)用镊子夹取蜡模在酒精灯上稍加热,并倒入少许从温箱中取出的纯石蜡。(2)再将镊子在酒精灯上稍加热,夹取材料将切面朝下放入蜡模中,排列整齐,再放上包埋盒,轻轻倒入熔蜡。5、切片、展片、贴片(1)将包埋好的石蜡块固定在切片机上,调整厚度调节器到所需的切片厚度,一般为4~6μm。(2)切下的组织薄片要放到加热的水中烫平,再贴到载玻片上,放45℃恒温箱中烘干。二、HE染色1、脱蜡、复水(1)保持水浴锅温度为60℃,将切片放入干燥的染色缸内,放入水浴锅中,30min至蜡熔化。(2)石蜡切片经二甲苯Ⅰ、Ⅱ脱蜡各5min,然后放入100%、95%、90%、80%、70%各级酒精溶液中各3~5min,再放入蒸馏水中3min,以便染料可以进入组织。2、染色、脱水(1)切片放入苏木精中染色约10~30min,用流水冲洗约15min,使切片颜色变蓝。(2)将切片放入1%盐酸乙醇液中褪色,几秒后见切片变红、颜色较浅即可,后将切片再放入流水中使其恢复蓝色。(3)切片放入50%、70%、80%乙醇中各3~5min。慢病毒载体包含了包装、转染、稳定整合所需要的遗传信息。海南外包科研技术服务实验
细胞核是细胞的控制中心,它包含了遗传物质DNA,控制着细胞的生长和分裂。浙江血液科研技术服务购买
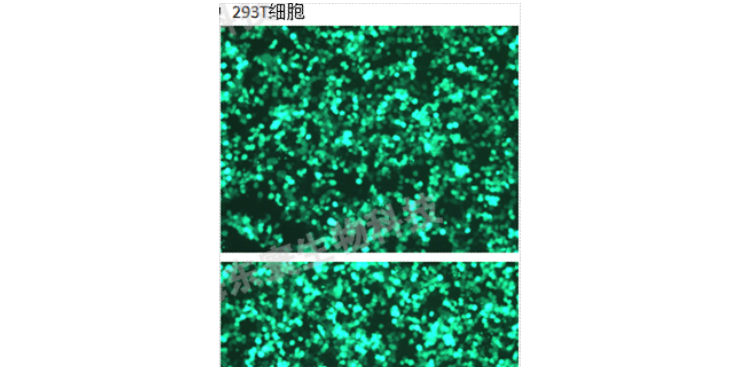
采用opti-MEM和Lipo3000分别转染含有目的基因的pMSCV-eGFP、VSV、GAG质粒及对照载体,每皿加入脂质体-质粒转染混悬液按购买脂质体相关说明书操作定量。继续培养24h。2)24小时后,将培养基更换为新鲜的DMEM完全培养基,放进细胞培养箱继续培养48~72h。3)48~72h后收集上层培养液,并过μm滤膜,采用ELISA法对所获得的慢载体进行滴度测定。如不及时使用可以冻存于-80℃。3、慢转染1)转染前1天将细胞接种6孔培养板,时细胞的融合率约为50%,前需换液,加入1mLDMEM完全培养基。2)冰浴融化后加入相应体积的液及聚凝胺(Polybrene),混匀后放入37℃孵箱中继续培养3)4h后补充1mL培养基,14h后换液(24h内换液即可)。4)72h后用倒置显微镜观察荧光,监测效率,出现较多荧光时将等量的转染细胞和未转染细胞分别加入等浓度Puromycin(Puromycin或其他筛选浓度需要事先摸索)。5)待未转染细胞全部死亡并且可观察到满意荧光量时,降低Puromycin浓度培养。也可以挑去单克隆细胞株进行进一步培养,以得到满意的稳定表达目的基因的细胞株。6)使用qRT-PCR和Westernblot的方法检测目的基因的表达量和蛋白水平是否显著提高。7)由此可得三组细胞株:a.正常细胞株;b.空载载体的细胞株。浙江血液科研技术服务购买