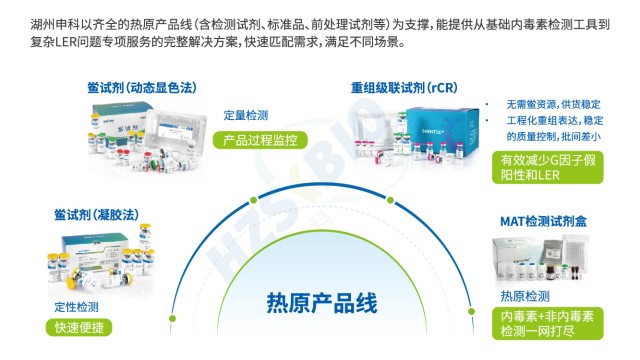
SHENTEK®重组级联试剂为内毒素检测高效解决方案。法规层面,符合美国药典(USP)、欧洲药典(EP)及日本药典(JP)要求,满足国际标准检测需求。抗干扰性强,与天然鲎具有相同反应机制,检测结果等效,适配复杂样本场景。特异性表现突出,不含 G 因子,避免与 β - D 葡聚糖反应产生假阳性,保障结果可靠。检测灵敏度高,线性范围达 0.005 - 5EU/mL ,可准确捕捉低浓度内毒素。反应快速,60分钟内即可完成检测,提升检测效率。兼容性佳,能兼容动态显色法的酶标仪,无需额外投入设备成本。关键的是,无动物源试剂,遵循 3R 原则(减少、替代、优化),不依赖鲎血,解决资源依赖问题,实现长期稳定供应。且通过重组表达生产,批次差异小,重复性好,稳定性高,为生物制品、制药等行业内毒素检测提供可靠、可持续的工具,助力企业把控质量,契合绿色环保与高效检测的双重需求,在保障检测准确性与合规性的同时,推动行业向更环保、更高效方向发展。
绘制内毒素标准曲线时,起始浓度需统一为 1000EU/mL,避免稀释误差。北京重组蛋白内毒素检测方法验证
内毒素检测方法易受样品基质干扰,法规要求在方法应用前必须进行干扰验证,选择标准曲线中点或一个靠近中点的内毒素浓度,作为供试品干扰试验种添加的内毒素浓度计算回收率,若回收率在 50%-200% 范围内,表明无明显干扰;若回收率异常,需通过过滤、中和、透析、加热处理等方式优化样品前处理。方法学确认还需涵盖线性范围(如 0.005-5 EU/mL)、精密度(批内 CV≤15%)、检测限(LOD≤0.01 EU/mL)等指标,确保方法在实际样品检测中稳定可靠,符合《美国药典》 <85>、《中国药典》通则 1143 等药典要求。
江苏医疗器械内毒素检测结果判定内毒素干扰试验回收率需在 50%-200%,验证样品基质不影响内毒素检出。
针对单核细胞活化反应测定法(MAT)通过检测内毒素的生物活性,有效规避低内毒素回收(LER)对內毒素检测的影响。其原理是:热原(包括被掩蔽的 LPS)活化单核细胞表面的 TLR 受体,释放 IL-6 等细胞因子,通过 ELISA 检测 IL-6 浓度,结合标准曲线推算热原含量。即使 LPS 因 LER 改变超分子结构,只要仍具生物活性,就能被 MAT 法识别。这种 “检测活性而非结构” 的特性,使 MAT 法成为 LER 场景下内毒素检测的重要补充,与其他方法协同构建高效可靠的热原防控体系。
重组试剂(rCR、rFC)是解决 LER 的重要工具,优化内毒素检测性能。重组鲎试剂(rCR)通过基因工程表达 C、B 因子及凝固酶原,剔除 G 因子,完全模拟天然鲎级联反应,灵敏度达 0.005EU/mL,与天然方法桥接容易,能避免 LPS 结构变化导致的假阴性;重组 C 因子(rFC)灵敏度 0.005-5EU/mL,性状稳定、均一性好,虽需荧光酶标仪,但对部分 LER 场景(如无蛋白质干扰)适配性强。二者摆脱了天然鲎试剂的局限,为内毒素检测提供更抗 LER 的选择,契合行业技术趋势。
内毒素检测中,脂多糖(LPS) 聚集体过度变小(近单体)可能降低检测信号。
生物制品(如单抗、疫苗、重组蛋白)注射剂因直接进入人体,对细菌内毒素残留限值要求严苛(通常≤0.5 EU/mg 或更低),检测面临基质复杂、干扰物质多等挑战。样品中常见的蛋白质、螯合剂、表面活性剂等可能抑制或增强 LAL 反应,需通过预处理消除干扰:如采用稀释法降低基质浓度、添加中和剂(如 Mg²⁺)修复反应体系,或使用热灭活去除蛋白类干扰物。此外,生物制品生产全流程需进行内毒素监控,从细胞培养上清、纯化中间品到终产品均需检测,确保工艺去除内毒素的有效性,符合 ICH Q6B 等法规对 “关键质量属性” 的控制要求。
70% 葡萄糖等高渗溶液会使鲎试剂蛋白变性,检测前需用检查用水稀释样本。江苏医疗器械内毒素检测结果判定
天然鲎试剂依赖鲎血,资源短缺,重组试剂成内毒素检测未来趋势。北京重组蛋白内毒素检测方法验证
内毒素检测方法验证需覆盖多项参数,确保方法可靠:线性范围需包含样品预期浓度(如 0.01-10 EU/mL),相关系数 R²≥0.98;准确度通过加标回收率评估,应在 50%-200% 范围内;精密度包括批内和批间精密度,CV 值均应≤15%;检测限(LOD)需低于产品限值的 1/2(如限值 0.5 EU/mL,LOD 应≤0.25 EU/mL);专属性需证明无干扰物质影响(如 β- 葡聚糖、蛋白质不引发假阳性)。验证通过后,方法需经实验室负责人批准方可使用,且定期需进行一次回顾性验证,确认方法持续有效。
北京重组蛋白内毒素检测方法验证