-
 太仓INDeCTD报价04.20
太仓INDeCTD报价04.20紧急申报与特殊通道:FDA设置紧急申报通道(如Pre-EUA和EUA),允许在公共卫生事件中快速提交资料。此类申请需在模块1.19注明特殊标识,并通过ESG加急处理。 eCTD版本兼容性与过渡策略:e...

-
 浙江ANDAeCTD品牌04.20
浙江ANDAeCTD品牌04.20区域化差异与多国协作挑战 欧盟eCTD需兼容成员国特定要求,例如模块一的行政信息需符合各国语言和法规差异。互认程序(MRP)中,参考成员国(RMS)的评估报告需被其他成员国认可,若出现分歧需由CMDh...
-
 徐汇区国内注册eCTD格式04.20
徐汇区国内注册eCTD格式04.20美国于2003年成为全球早采用eCTD(电子通用技术文档)的国家之一,初由CDER和CBER作为电子提交平台试点。2008年起,eCTD正式成为药申请(NDA)和生物制品许可申请(BLA)的标准格式,...
-
 静安区中国eCTD服务商04.20
静安区中国eCTD服务商04.20eCTD验证标准的严格性与分类:欧盟对eCTD的验证要求分为“错误”“警告”和“提示信息”三级,其中“错误”项直接导致申报被拒。验证项目涵盖六大类共149条,包括文件命名规范(如路径长度限制)、PDF...
-
 芜湖CDE eCTD性价比04.19
芜湖CDE eCTD性价比04.19eCTD的实施为监管机构和企业带来了多重机遇。电子化申报资料能够极大地加速审评效率,减少人为判断错误和数据混淆的情况,从而提高审评的准确性和速度。同时,eCTD带来的数据标准化机遇使得全球监管机构的资...
-
 闵行区eCTD名称04.19
闵行区eCTD名称04.19多国审评程序与eCTD递交途径的适配:欧盟药品审评程序包括集中(CP)、分散(DCP)、互认(MRP)和国家程序(NP),eCTD需适配不同程序的递交要求。例如: 集中审评程序(CP):通过EMA...
-
 杭州eCTD报价04.19
杭州eCTD报价04.19美国eCTD的强制实施时间与范围:美国自2017年5月5日起要求药申请(NDA)、仿制药申请(ANDA)和生物制品许可申请(BLA)必须通过eCTD格式提交,2018年5月5日进一步扩展至临床试验申请...
-
 江西ANDAeCTD04.19
江西ANDAeCTD04.19欧洲药品管理局:集中审评程序由欧洲药品管理局(European Medicines Agency, EMA)负责协调。 人用药品委员会:人用药品委员会(Committee for Medicinal ...
-
 静安区NDAeCTD服务商04.19
静安区NDAeCTD服务商04.19审评效率与时间线优化 eCTD的标准化缩短了审评周期:集中程序平均审评时间从18个月降至12个月,互认程序可在90天内完成成员国意见协调。自动化验证工具减少了格式错误导致的退审率,但复杂药学数据的科学...
-
 湖北eCTD哪个品牌好04.18
湖北eCTD哪个品牌好04.18美国eCTD验证采用三级分类:“错误”(必须修正)、“警告”(建议修正)、“提示信息”(参考)。例如,PDF文件版本不符或加密保护属于“错误”,而书签路径非相对性则可能列为“警告”。验证失败将直接导致...
-
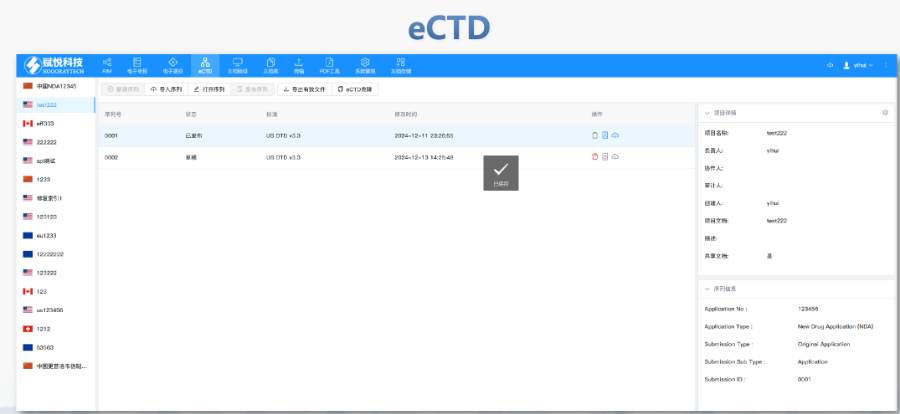 浦东新区国产eCTD发布系统04.18
浦东新区国产eCTD发布系统04.18eCTD在欧盟药品监管中的历史背景:欧盟eCTD(电子通用技术文档)的发展始于对临床试验和药品审评流程标准化的需求。2001年,欧盟引入《临床试验指令》(CTD)作为统一的法律框架,但其分散的成员国申...
-
浙江赋悦科技eCTD发布软件04.18
澳大利亚的药品电子通用技术文档(eCTD)注册申报体系是澳大利亚y药品商品管理局(TGA)推动药品审评现代化的重要举措。eCTD作为国际通行的电子化注册申报标准,通过结构化数据格式(如XML)整合了药...