-
 高新区化学药品eCTD服务放心可靠04.13
高新区化学药品eCTD服务放心可靠04.13欧美eCTD实施经验丰富,中国可借鉴以加速进程。中国可能会经历从企业自愿eCTD提交到强制eCTD提交的过渡,且将紧随ICH步伐,尤其在CMC资料整理方面。全球正向eCTD 4.0过渡,中国也不例外,...

-
 上海电子申报eCTD服务价格04.13
上海电子申报eCTD服务价格04.13多国审评程序与eCTD递交途径的适配:欧盟药品审评程序包括集中(CP)、分散(DCP)、互认(MRP)和国家程序(NP),eCTD需适配不同程序的递交要求。例如: 集中审评程序(CP):通过EMA...
-
 吴江区国际注册eCTD欢迎选购04.12
吴江区国际注册eCTD欢迎选购04.12ANDA一般不需要提供临床前(动物)和临床(人体)数据来证明其安全性和有效性(即免毒理和临床),作为替代,申请人必须合理证明其产品与原研药相比是生物等效的。 按照《联邦食品、药品和化妆品法》第 505...
-
 闵行区CDE eCTD销售电话04.12
闵行区CDE eCTD销售电话04.12申报流程与要求 资料准备 内容要求:包括产品描述、生产工艺(原材料来源、设备参数等)、质量控制标准(SOP、稳定性数据)、安全性与毒性研究等。 格式规范: 采用CTD(通用技术文件)格式,按模块...
-
 合肥生物制品eCTD供应商04.12
合肥生物制品eCTD供应商04.12经济影响与成本效益 尽管初期投入较高(平均每企业需50万欧元),但eCTD可减少30%的审评延迟成本,长期效益。仿制药企业通过eCTD复用原研数据,节省80%的申报准备时间。欧盟预算拨款2亿欧元资助中...
-
南京药品注册eCTD递交04.12
eCTD在欧盟药品监管中的历史背景:欧盟eCTD(电子通用技术文档)的发展始于对临床试验和药品审评流程标准化的需求。2001年,欧盟引入《临床试验指令》(CTD)作为统一的法律框架,但其分散的成员国申...
-
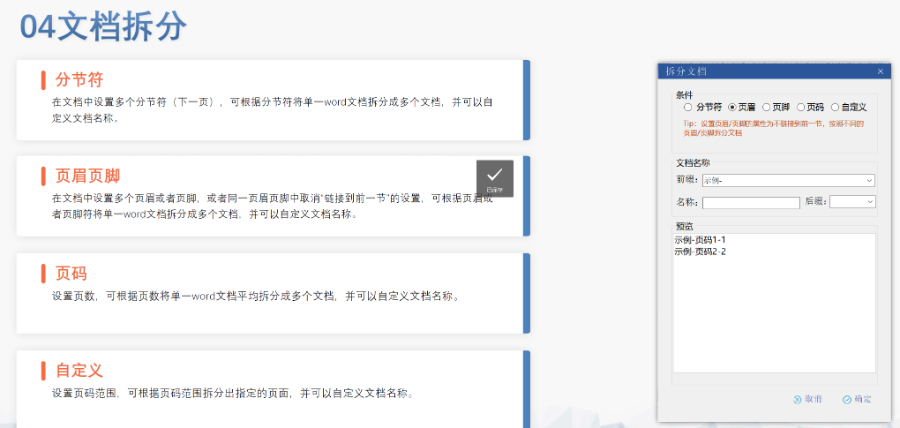 苏州新药eCTD注册系统04.12
苏州新药eCTD注册系统04.12eCTD验证标准的严格性与分类:欧盟对eCTD的验证要求分为“错误”“警告”和“提示信息”三级,其中“错误”项直接导致申报被拒。验证项目涵盖六大类共149条,包括文件命名规范(如路径长度限制)、PDF...
-
安徽电子申报eCTD服务商04.11
澳大利亚的药品电子通用技术文档(eCTD)注册申报体系是澳大利亚y药品商品管理局(TGA)推动药品审评现代化的重要举措。eCTD作为国际通行的电子化注册申报标准,通过结构化数据格式(如XML)整合了药...
-
 工业园区化学药品eCTD性价比04.11
工业园区化学药品eCTD性价比04.11设施费动态调整 API工厂和制剂工厂年费分别约6.8万和14.5万美元(2025财年),CMO工厂费用为制剂费的24%。国外工厂需额外支付1.5万美元跨境检查费。 缴费时限与惩罚 费用需在财年首日(...
-
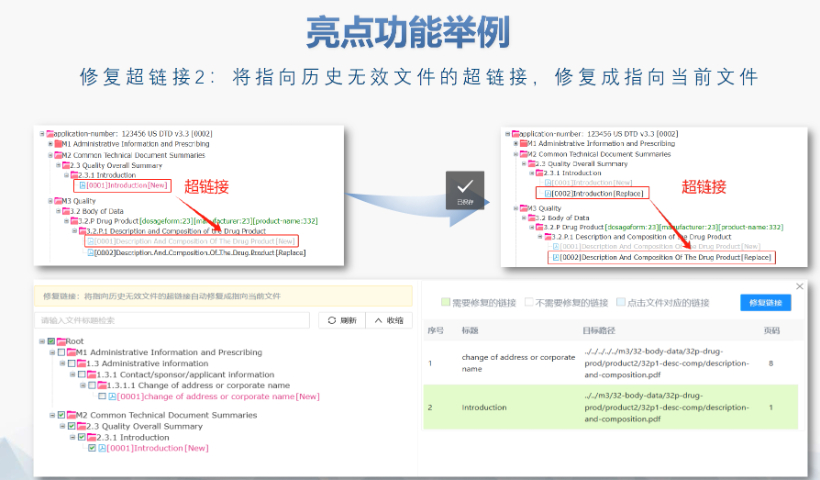 吴江区国产eCTD服务介绍04.11
吴江区国产eCTD服务介绍04.11文件生命周期管理:eCTD支持文件替换(Replace)、删除(Delete)等操作,而非增文件。例如,更临床研究方案时需用Replace操作覆盖旧版本。基线提交(Baseline Submissio...
-
 浙江原料药eCTD品牌04.11
浙江原料药eCTD品牌04.11eCTD的技术架构与模块要求:美国eCTD基于XML技术,严格遵循ICH M4框架,分为5个模块:模块1(地区行政信息)、模块2(技术总结)、模块3-5(质量、非临床与临床数据)。其中,模块1需包含F...
-
徐汇区中国eCTD常用解决方案04.11
美国于2003年成为全球早采用eCTD(电子通用技术文档)的国家之一,初由CDER和CBER作为电子提交平台试点。2008年起,eCTD正式成为药申请(NDA)和生物制品许可申请(BLA)的标准格式,...