-
 山西eCTD报价04.06
山西eCTD报价04.06欧盟eCTD的递交途径与技术要求 不同审评程序对应不同递交渠道:集中程序(CP)通过EMA的eSubmission Gateway或Web Client提交,分散程序(DCP)和互认程序(MRP)则需...

-
 吴江区药品注册eCTD格式04.05
吴江区药品注册eCTD格式04.05申报流程与要求 资料准备 内容要求:包括产品描述、生产工艺(原材料来源、设备参数等)、质量控制标准(SOP、稳定性数据)、安全性与毒性研究等。 格式规范: 采用CTD(通用技术文件)格式,按模块...
-
 芜湖CDE eCTD哪家好04.05
芜湖CDE eCTD哪家好04.05eCTD生命周期管理与变更提交:欧盟要求eCTD申报资料覆盖药品全生命周期,包括提交、补充申请及实质性变更。例如,增成员国需提交“附加成员国序列”,审评时间约52-83天;重大变更(如生产工艺调整)需...
-
 杨浦区仿制药eCTD性价比04.05
杨浦区仿制药eCTD性价比04.05eCTD 4.0版本的过渡与升级:FDA于2023年启动eCTD 4.0技术试点,2024年9月正式接收申请,计划2029年完成全过渡。4.0版本改用HL7 RPS标准替代XML,支持双向通信和跨申请...
-
 山东原料药eCTD服务价格04.05
山东原料药eCTD服务价格04.05多国审评程序与eCTD递交途径的适配:欧盟药品审评程序包括集中(CP)、分散(DCP)、互认(MRP)和国家程序(NP),eCTD需适配不同程序的递交要求。例如: 集中审评程序(CP):通过EMA...
-
 苏州国内注册eCTD递交04.05
苏州国内注册eCTD递交04.05eCTD的技术架构与模块要求:美国eCTD基于XML技术,严格遵循ICH M4框架,分为5个模块:模块1(地区行政信息)、模块2(技术总结)、模块3-5(质量、非临床与临床数据)。其中,模块1需包含F...
-
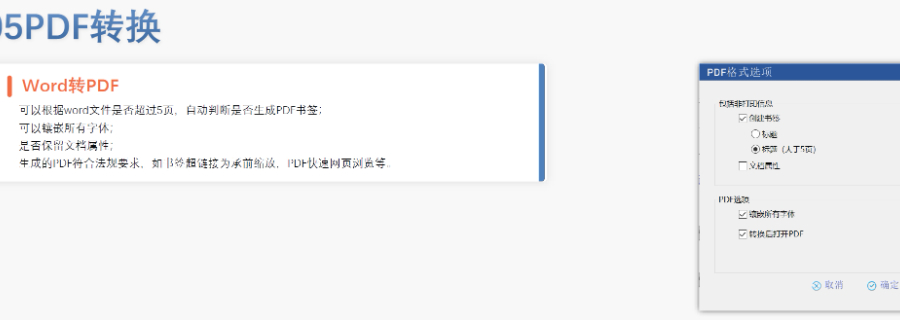 太仓ANDAeCTD报价04.04
太仓ANDAeCTD报价04.04DMF维护与合规 年度更 即使无变更,每年需提交声明;重大工艺/设施变更需及时通知客户并更文件。 现场检查 原料药企业需通过FDA现场检查,验证是否符合ICH Q7 GMP标准,并与DMF内容一致...
-
 合肥化学药品eCTD性价比高04.04
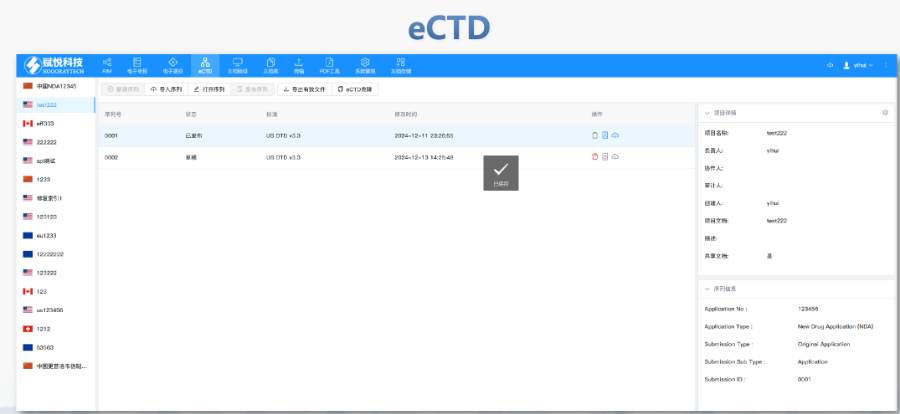
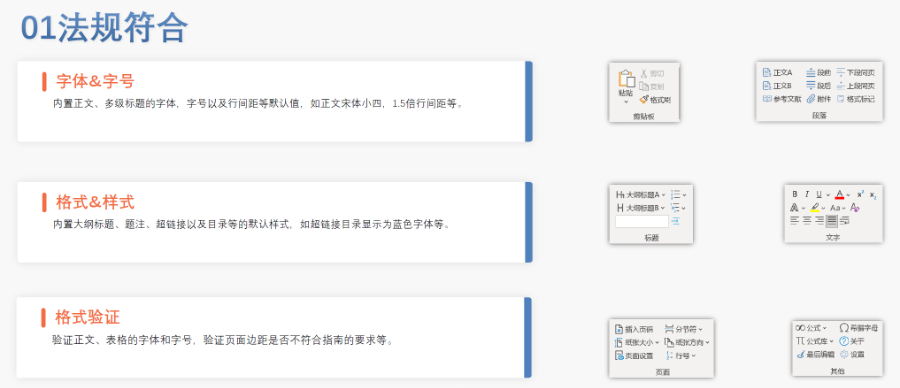
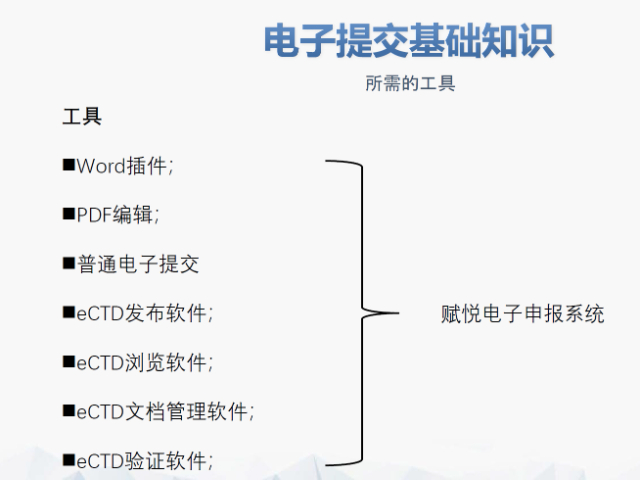
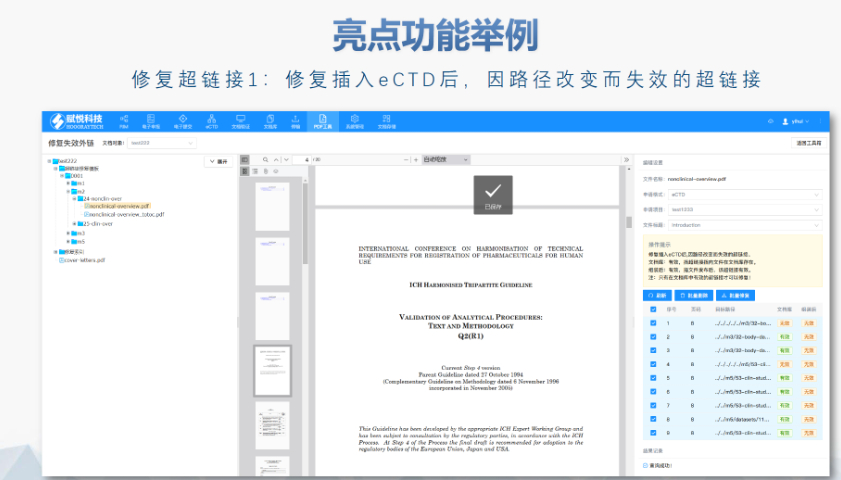
合肥化学药品eCTD性价比高04.04赋悦eCTD系统 文件验证与修复 支持自动验证文件格式(如PDF属性、字体嵌入、超链接完整性等),并一键修复不符合法规要求的文件。例如,系统会自动检查XML主干文件的结构合规性,确保符合中国、美国、...
-
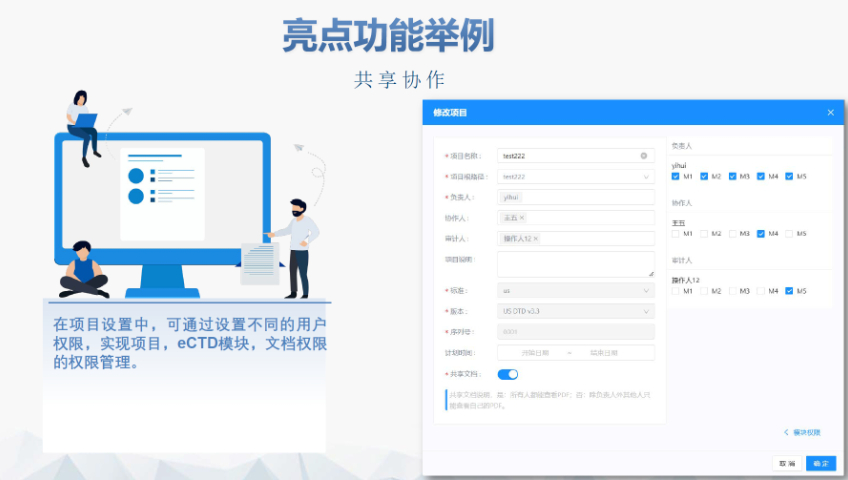 太仓原料药eCTD报价04.04
太仓原料药eCTD报价04.04电子签章与安全性 FDA要求所有PDF文件需经数字签名,并通过MD5校验确保传输完整性。签章需符合21 CFR Part 11的电子记录规范,部分情况下允许临时放宽(如期间的远程签署)。 多模块协同...
-
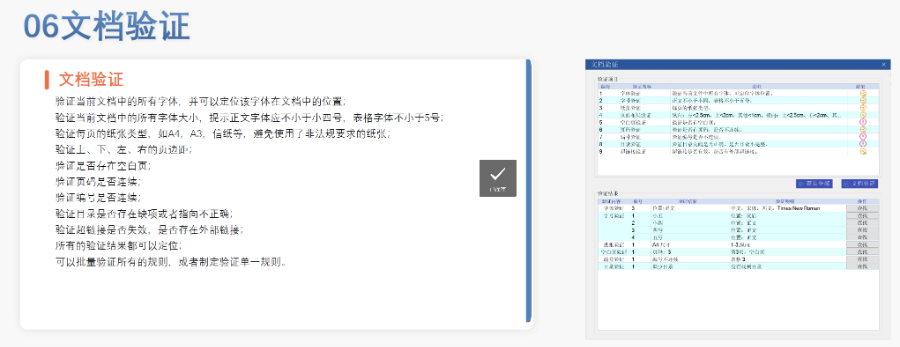 杭州国际注册eCTD递交04.04
杭州国际注册eCTD递交04.04生命周期管理与变更递交 eCTD支持全生命周期管理,申请人需通过序列更(Sequence)反映药品变更信息。例如CEP证书的更需提交“变更说明表”,对比已批准和拟修改内容,并附修订版技术文档。重大变更...
-
中国eCTD服务介绍04.04
生命周期管理与变更递交 eCTD支持全生命周期管理,申请人需通过序列更(Sequence)反映药品变更信息。例如CEP证书的更需提交“变更说明表”,对比已批准和拟修改内容,并附修订版技术文档。重大变更...
-
宁波NDAeCTD发布软件04.03
澳大利亚的药品电子通用技术文档(eCTD)注册申报体系是澳大利亚y药品商品管理局(TGA)推动药品审评现代化的重要举措。eCTD作为国际通行的电子化注册申报标准,通过结构化数据格式(如XML)整合了药...